ڈء‰»ٹيٹàژ،—أ‚جچLڈê GIcancer-net



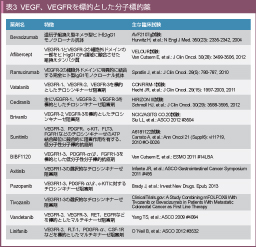
پ@ٹà‚ج“ء’¥‚ج1‚آ‚ةپuinducing angiogenesisپv‚ھ‚ ‚éپB‘ه’°ٹà‚جŒŒٹاگVگ¶‚ة‚حپAٹàچ×–E‚إ‚جŒŒٹا“à”çچ×–E‘گBˆِژq (vascular endothelial growth factor: VEGF) ژYگ¶‚ئŒŒٹا“à”çچ×–E‚جVEGFR‚ة‚و‚éƒIپ[ƒgƒNƒ‰ƒCƒ“ (ژ©Œب•ھ”ه) ‚ھ‘ه‚«‚ٹض—^‚µ‚ؤ‚¢‚éپB‚ـ‚½پAVEGF‚âVEGFR‘jٹQچـ‚حپAŒŒٹاگVگ¶‚ج—}گ§‚ج‚ف‚ب‚炸پAژîل‡ŒŒٹا‚جگ³ڈي‰»چى—p‚ة‚و‚èچ×–EڈٹQگ«چRٹàچـ‚ج‘gگD“àˆعچs‚ًڈمڈ¸‚³‚¹‚邱‚ئ‚إ•¹—pŒّ‰ت‚ً”ٹِ‚·‚é‚ئچl‚¦‚ç‚ê‚ؤ‚¢‚éپB
پ@Bevacizumab‚حVEGF‚ة“ءˆظ“I‚ةŒ‹چ‡‚·‚éˆâ“`ژq‘gٹ·‚¦Œ^‚جƒLƒپƒ‰Œ^ƒqƒg‰»IgG1ƒ‚ƒmƒNƒچپ[ƒiƒ‹چR‘ج (•ھژq—ت–ٌ149,000) ‚إ‚ ‚éپB“ïژ،گ«ŒإŒ`ٹà‚ً‘خڈغ‚ةBevacizumab‚ًday 0پA28پA35پA42‚ة“ٹ—^‚µˆہ‘Sگ«‚ئ–ٍ•¨“®‘ش‚ًŒں“¢‚·‚é‘وI‘ٹژژŒ±‚ھچs‚ي‚ꂽŒ‹‰تپA“ٹ—^—ت0.3mg/kg‚©‚ç10mg/kg‚ج”حˆح‚إ‚حگüŒ`گ«‚ج–ٍ•¨“®‘ش‚ًژ¦‚µپA”¼Œ¸ٹْ‚ح–ٌ21“ْ‚إ‚ ‚ء‚½7)پB‚ـ‚½پAŒŒ’†‚جfree VEGF‚حBevacizumab“ٹ—^—ت0.3mg/kgˆبڈم‚إ‘ھ’èٹ´“xˆب‰؛‚ئ‚ب‚ء‚½پB—LٹQژ–ڈغ‚ئ‚µ‚ؤ‚حŒŒˆ³ڈمڈ¸ (10پ`15 mmHg)پAgrade 1/2‚ج“ھ’ةپAڑq‹C‚ب‚ا‚ً”F‚كپA3.0mg/kgˆبڈم‚ج“ٹ—^—ت‚إژîل‡“àڈoŒŒ‚ھ5—ل’†2—ل”F‚ك‚ç‚ꂽ‚ھپAچإ‘ه‘د—ت (maximum tolerated dose: MTD) ‚ة‚ح’B‚µ‚ب‚©‚ء‚½پB‘O—صڈ°ژژŒ±‚ة‚¨‚¢‚ؤپA’èڈيڈَ‘ش‚جƒgƒ‰ƒt’l‚ھ10-30ƒتg/mLˆبڈم‚جڈêچ‡‚إ‚جچRژîل‡Œّ‰ت‚ھچإ‘ه‚إ‚ ‚ء‚½‚±‚ئ‚ھچl—¶‚³‚êپA2پ`3mg/kgڈT‚P‰ٌ“ٹ—^‚ھگ„ڈ§—p—ت (recommended dose: RD) ‚ئ‚³‚ꂽپB
پ@‘ه’°ٹà‚ًٹـ‚قŒإŒ`ٹàٹ³ژز‚ً‘خڈغ‚ةBevacizumab (3mg/kgڈT1‰ٌ“ٹ—^) ‚ج•¹—p—أ–@‚ًŒں“¢‚µ‚½‘وIb‘ٹژژŒ±‚إ‚حپA‘ه’°ٹàٹ³ژز‚ة‘خ‚·‚é5-FU/LV—أ–@‚ئ‚ج•¹—p—أ–@‚إ‘ه‚«‚ب“إگ«‚ً”F‚ك‚ب‚©‚ء‚½8)پB‚»‚µ‚ؤپA5-FU/LV—أ–@‚ئ5-FU/LV + Bevacizumab (5mg/kgٹuڈT“ٹ—^) —أ–@پA5-FU/LV + Bevacizumab (10mg/kgٹuڈT“ٹ—^) —أ–@‚ً”نٹr‚·‚é–³چىˆ×‰»‘وII‘ٹژژŒ± (AVF0780gژژŒ±) ‚إ‚حپA‘tŒّ—¦‚ح‚»‚ꂼ‚ê17%پA40%پA24%پAPFS (progression-free survival) ’†‰›’l‚ح‚»‚ꂼ‚ê5.2ƒ•ŒژپA9.0ƒ•ŒژپA7.2ƒ•ŒژپAOS (overall survival) ’†‰›’l‚ح‚»‚ꂼ‚ê13.8ƒ•ŒژپA21.5ƒ•ŒژپA16.1ƒ•Œژ‚ئپA‚¢‚¸‚ê‚à5mg/kg•¹—pŒQ‚إ—اچD‚بگ¬گر‚إ‚ ‚ء‚½9)پB‚±‚جŒ‹‰ت‚©‚çپAˆبŒم‚ج‘ه’°ٹàڈ‰‰ٌژ،—أ—ل‚ً‘خڈغ‚ة‚µ‚½—صڈ°ژژŒ±‚إ‚حپA5mg/kgٹuڈT“ٹ—^‚ج“ٹ—^—ت‚ھچج—p‚³‚ê‚ؤ‚¢‚éپB“–ژ‚ج•Wڈ€ژ،—أ‚إ‚ ‚ء‚½IFL—أ–@‚ئBevacizumab•¹—p—أ–@‚ً”نٹr‚·‚é‘وIII‘ٹژژŒ± (AVF2107gژژŒ±) ‚إ‚حپA“–ڈ‰IFL + placeboŒQپAIFL + BevacizumabŒQپA5-FU/LV + BevacizumabŒQ‚ج3ŒQ‚ة1:1:1‚إٹ„•t‚¯‚ç‚ꂽ‚ھپA313—ل‚ھ“oک^‚³‚ꂽژ“_‚ج’†ٹش‰ًگح‚ة‚¨‚¢‚ؤIFL + BevacizumabŒQ‚جˆہ‘Sگ«‚ةڈd‘ه‚ب–â‘è‚ھ‚ب‚©‚ء‚½‚±‚ئ‚©‚çپAˆبŒم‚حIFL پ} Bevacizumab‚ج”نٹrژژŒ±‚ئ‚µ‚ؤŒp‘±‚³‚êپAچإڈI“I‚ةIFL + placeboŒQ411—لپAIFL + BevacizumabŒQ402—ل‚ھ“oک^‚³‚ꂽ10)پBŒ‹‰تپAژه—v•]‰؟چ€–ع‚إ‚ ‚éOS‚ة‚¨‚¢‚ؤIFL + placeboŒQ15.6ƒ•ŒژپAIFL + BevacizumabŒQ20.3ƒ•Œژ‚ئپABevacizumab•¹—p‚إ—Lˆس‚ب‰„’·‚ً”F‚ك‚½ (p<0.001)پB‚»‚جŒمپABICC-CژژŒ±پANO16966ژژŒ±‚ھچs‚ي‚êپAL-OHP‚âFOLFIRI‚ض‚جBevacizumab‚ج—L—pگ«‚ھژ¦‚³‚ê‚ؤ‚¢‚é11, 12)پB
پ@‚ـ‚½پAٹùژ،—أ—ل‚إ‚حپACPT-11 + 5-FU•¹—p—أ–@•s‰—ل‚ً‘خڈغ‚ةپAFOLFOX4—أ–@پAFOLFOX4 + Bevacizumab—أ–@پABevacizumab’P“ئ—أ–@‚ً”نٹr‚·‚é‘وIII‘ٹژژŒ± (E3200ژژŒ±) ‚ھچs‚ي‚êپABevacizumab•¹—p‚ة‚و‚éPFSپAOS‚ج‰„’·‚ھژ¦‚³‚ꂽ13)پB–{ژژŒ±‚إ‚حپA‘O—صڈ°‚جŒ‹‰ت‚إ—p—تˆث‘¶گ«‚ةBevacizumab‚جŒّ‰ت‚ھ‘‹‚·‚邱‚ئپA”ٌڈ¬چ×–E”xٹà‚ة‚¨‚¯‚éCarboplatin + Paclitaxel (CP) —أ–@‚ئCP + Bevacizumab (7.5mg/kg 3ڈT–ˆ) —أ–@پACP + Bevacizumab (15mg/kg 3ڈT–ˆ) —أ–@‚ج–³چىˆ×‰»”نٹr‘وII‘ٹژژŒ±‚إپA15mg/kg•¹—pŒQ‚ھ7.5mg/kg•¹—pŒQ‚ئ”نٹr‚µ‚ؤ‘tŒّ—¦پATTP (time to progression) ‚ھ—اچD‚بŒXŒü‚إ‚ ‚ء‚½14) ‚±‚ئ‚ب‚ا‚ھچl—¶‚³‚êپABevacizumab‚ج“ٹ—^—ت‚ح10mg/kg ٹuڈT‚ئ‚³‚ê‚ؤ‚¢‚éپBˆبڈم‚جŒ‹‰ت‚ً“¥‚ـ‚¦‚ؤپA•ؤچ‘FDA‚ح2006”N6Œژ‚ة2nd-line‚ة‚¨‚¢‚ؤ5-FU/LV‚ًٹـ‚ق‘S‚ؤ‚جƒŒƒWƒپƒ“‚إBevacizumab•¹—p—أ–@‚ًڈ³”F‚µ‚½پB
پ@‘ه’°ٹà‚ة‘خ‚·‚éBevacizumab•¹—p—أ–@‚ج—صڈ°“I‚ب“ءگ«‚ئ‚µ‚ؤپA’Pچـ‚إ‚ح—LŒّگ«‚ھ–R‚µ‚¢‚à‚ج‚جپAچ×–EڈٹQگ«چRٹàچـ‚ئ‚ج•¹—p‚ة‚و‚èچRژîل‡Œّ‰ت‚ً”ٹِ‚µپA‚©‚آچ×–EڈٹQگ«چRٹàچـ‚ة‚و‚é‰؛—ںپAˆ«گS‚ب‚ا‚ج•›چى—p‚ً‘‹‚µ‚ب‚¢‚±‚ئ‚ھ‹“‚°‚ç‚ê‚éپBژہچغپANO16966ژژŒ±‚ة‚¨‚¯‚é–ٍچـ“ٹ—^—ت‚جŒں“¢‚ة‚¨‚¢‚ؤپAچ×–EڈٹQگ«چRٹàچـ‚ج—\’è“ٹ—^—ت‚ة‘خ‚·‚éژہچغ‚ج“ٹ—^—ت (‘ٹ‘خ—p—ت‹“xپArelative dose intensity: RDI) ‚حپABevacizumab•¹—pŒQ‚ئplaceboŒQ‚ئ‚إچ·‚ً”F‚ك‚ب‚©‚ء‚½12)پBچ×–EڈٹQگ«چRٹàچـ‚ئ‚ج•¹—p—أ–@‚ة‚ؤژ،—أŒّ‰ت‚جڈمڈو‚¹‚ًٹْ‘ز‚·‚é‚ة‚حپAƒxپ[ƒX‚جژ،—أ‚جRDI‚ً•غ‚آ‚±‚ئ‚ھڈd—v‚ب—v‘f‚ج1‚آ‚إ‚ ‚é‚ئچl‚¦‚ç‚ê‚éپB
پ@Aflibercept‚حVEGFR-1‚ئVEGFR-2‚جچ×–EٹOƒhƒپƒCƒ“‚جˆê•”‚ًƒqƒgIgG1‚جFc—جˆو‚ة—Zچ‡‚³‚¹‚½‘gٹ·‚¦ƒ^ƒ“ƒpƒNژ؟‚إ‚ ‚èپAVEGF‚ة‘خ‚·‚éچR‘جˆم–ٍ‚إ‚ح‚ب‚ (ƒfƒRƒC) ƒŒƒZƒvƒ^پ[‚إ‚ ‚éپBAflibercept‚حVEGF (VEGF-A) ‚ج‘S‚ؤ‚جˆظگ«‘ج‚¾‚¯‚إ‚ب‚پA‘¼‚جVEGFRƒٹƒKƒ“ƒh‚إ‚ ‚éVEGF-BپAPlGF‚ة‚àŒ‹چ‡ (trap) ‚µپAٹà‚جŒŒٹاگVگ¶‚ً‘jٹQ‚·‚邱‚ئ‚إچRژîل‡Œّ‰ت‚ً”ٹِ‚·‚éپB“ïژ،گ«ŒإŒ`ٹà‚¨‚و‚ر”ٌƒzƒWƒLƒ“ƒٹƒ“ƒpژî‚ً‘خڈغ‚ئ‚µ‚½Aflibercept’Pچـ—أ–@‚ج‘وI‘ٹ—p—ت‘Q‘ژژŒ±‚إ‚حپA“oک^‚³‚ꂽ47گl‚جٹ³ژز‚ة‘خ‚µ‚ؤ—p—ت0.3پ`7.0mg/kgپAٹuڈT‚إ“ٹ—^‚³‚ꂽ15)پBŒ‹‰تپA7.0mg/kgŒQ‚إ13—ل’†2—ل‚ةDLT (dose-limiting toxicity) (’¼’°’×ل‡پA’`”’”A) ‚ھ”F‚ك‚ç‚ꂽپB‚ـ‚½پAچ‚ŒŒˆ³‚ً1.0پ`3.0mg/kg‚إ‚ج“ٹ—^ٹْٹش“à‚ة14.3پ`16.7%پA4.0پ`7.0mg/kg‚إ‚ح57.1پ`75.0%‚ة”F‚ك‚½پB–ٍ•¨“®‘ش (pharmacokinetics: PK) /–ٍ—حٹw (pharmacodynamics: PD) ‰ًگح‚إ‚حپA—V—£Œ^VEGF trap‚ج”¼Œ¸ٹْ‚ح–ٌ5“ْ‚إ“ٹ—^—ت‚ةˆث‘¶‚µ‚ؤ”Z“xڈمڈ¸‚ھ‚ف‚ç‚ꂽ‚ھپAVEGFŒ‹چ‡Œ^VEGF trap‚ج”¼Œ¸ٹْ‚ح–ٌ20“ْ‚إ2.0mg/kgˆبڈم‚ج“ٹ—^—ت‚إ‚حƒvƒ‰ƒgپ[‚ة’B‚µ‚½پB‚±‚ج‚±‚ئ‚©‚çپAAflibercept‚ج“ٹ—^—ت‚ھ2.0kg/mgˆبڈم‚إ‚ ‚ê‚خŒŒ’†VEGF‚ًڈ\•ھtrap‚إ‚«‚ؤ‚¢‚é‚ئچl‚¦‚ç‚êپA’Pچـ—أ–@‚إ‚جگ„ڈ§“ٹ—^—ت‚ح4.0mg/kgٹuڈT“ٹ—^‚ئŒˆ’肳‚ꂽپB
پ@‚³‚ç‚ةŒإŒ`ٹà‚ً‘خڈغ‚ئ‚µ‚ؤCPT-11 + LV5FU2‚ةAflibercept‚ج•¹—p‚ًچs‚¤—p—ت‘Q‘‘و‡T‘ٹژژŒ±‚ھچs‚ي‚ê (2پ`6mg/kg)پA‘ه’°ٹà23—ل‚ًٹـ‚ق38—ل‚ھ“oک^‚³‚ꂽ16)پBDLT‚ح4mg/kgŒQ‚إ2 /12—ل (’`”’”A)پA5mg/kgŒQ‚إ2/10—ل (Œû“à‰ٹپAˆفگH“¹‹t—¬)پA6mg/kgŒQ‚إ3/12—ل (””Mگ«چD’†‹…Œ¸ڈڈاپAŒû“à‰ٹ) ‚ة”F‚ك‚½پBPK‰ًگح‚إ—V—£Œ^VEGF trap‚جŒŒ’†—ت‚ھڈيژŒ‹چ‡Œ^VEGF trap‚ًڈم‰ٌ‚é‚ج‚ح“ٹ—^—ت4mg/kgˆبڈم‚إ‚ ‚ء‚½‚±‚ئپA4mg/kgŒQ‚ج5 /10—ل‚إPR‚ھ”F‚ك‚ç‚ꂽ‚±‚ئپA‚»‚êˆبڈم‚ج—p—ت‘‰ء‚ة‚ؤ“إگ«‚âچRژîل‡Œّ‰ت‚ة–¾‚ç‚©‚بچ·‚ً”F‚ك‚ب‚©‚ء‚½‚±‚ئ“™‚©‚çپAگ„ڈ§“ٹ—^—ت‚ح4mg/kg‚ةŒˆ’肳‚ꂽپB‚³‚ç‚ةFOLFIRI—أ–@‚ئAflibercept (4mg/kg) •¹—p—أ–@‚جˆہ‘Sگ«ٹm”FژژŒ±‚ھ‘ه’°ٹà19—ل‚ًٹـ‚ق27—ل‚ً‘خڈغ‚ئ‚µ‚ؤچs‚ي‚êپAAfliberceptٹضکA‚ج—LٹQژ–ڈغ‚ئ‚µ‚ؤgrade 2/3‚جچ‚ŒŒˆ³‚ً‚»‚ꂼ‚ê26%پA15%پAgrade 2‚ج”گ¶چ¢“ï‚ً11%پAgrade 2‚ج’`”’”A‚ً4%”F‚ك‚é‚à”E—eگ«‚ح—اچD‚إ‚ ‚ء‚½‚±‚ئ‚©‚çپA‘وIII‘ٹژژŒ±‚جAflibercept‚جگ„ڈ§—p—ت‚ح4mg/kg‚ئ‚³‚ꂽپB
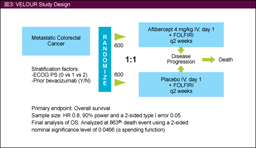
پ@‚»‚جŒمL-OHPƒxپ[ƒX‚جڈ‰‰ٌ‰»ٹw—أ–@‚ة•s‰‚ئ‚ب‚ء‚½‘ه’°ٹàٹ³ژز‚ً‘خڈغ‚ةFOLFIRI + placeboŒQ‚ئFOLFIRI + AfliberceptŒQ‚ئ‚ً”نٹr‚·‚é‘وIII‘ٹژژŒ± (VELOURژژŒ±) ‚ھچs‚ي‚ꂽ17)پBŒ‹‰تپAژه—v•]‰؟چ€–ع‚جOS’†‰›’l‚ح‚»‚ꂼ‚ê12.06ƒ•ŒژپA13.50ƒ•Œژ (HR=0.817, 95.34% CI: 0.713-0.937, p=0.0032)پA•›ژں•]‰؟چ€–ع‚جPFS’†‰›’l‚ح‚»‚ꂼ‚ê4.7ƒ•ŒژپA6.9ƒ•Œژ (HR=0.758, p<0.0001)پA‘tŒّ—¦‚ح‚»‚ꂼ‚ê11.1%پA19.8% (p<0.001) ‚ئپAAfliberceptŒQ‚إ—Lˆس‚ة—اچD‚إ‚ ‚ء‚½پB‚ـ‚½پA–{ژژŒ±‚حڈ‰‰ٌژ،—أ‚إ‚جBevacizumabژg—p—ً‚ھ‘w•تˆِژq‚ةٹـ‚ـ‚ê‚ؤ‚¨‚èپABevacizumabژg—p—ل‚ة‚¨‚¯‚é—LŒّگ«‚جŒں“¢‚إ‚حپABevacizumab–¢ژg—pŒQ853—ل‚إ‚حOS‚جHR‚ھ0.788پABevacizumabژg—pŒQ‚جHR‚ح0.862‚ئپAچRVEGFچR‘ج–ٍ‚إ‚ ‚éBevacizumab‚جژg—p—ً‚ھ‚ ‚éڈW’c‚ة‘خ‚µ‚ؤ‚à—اچD‚بŒXŒü‚ً”F‚ك‚½پBGrade 3/4‚ج—LٹQژ–ڈغ‚حپA‰؛—ںپAŒ‘‘سٹ´پAŒû“à‰ٹپAچ‚ŒŒˆ³پA• ’ةپAچD’†‹…Œ¸ڈپA’`”’”A‚ھAfliberceptŒQ‚إ‘½‚”F‚ك‚ç‚êپA—LٹQژ–ڈغ‚ة”؛‚¤ژ،—أ’†ژ~—ل‚حplaceboŒQ12.1%پAAfliberceptŒQ26.6%‚ئپAAfliberceptŒQ‚إ‘½‚©‚ء‚½پB‚ـ‚½پA5-FUپACPT-11‚جŒ¸—تپA’†ژ~‚ً—v‚µ‚½ٹ„چ‡‚حplaceboŒQ‚إ5-FU‚ھ21.7%پACPT-11‚ھ22.6%پAAfliberceptŒQ‚إ‚ح‚»‚ꂼ‚ê39.1%پA37.2%‚إ‚ ‚ء‚½پB
پ@ˆê•ûپAL-OHPƒxپ[ƒXƒŒƒWƒپƒ“‚ئAflibercept‚ج•¹—p‚ة‚آ‚¢‚ؤ‚حپAڈ‰‰ٌژ،—أ‚ة‚¨‚¯‚émFOLFOX6 پ} Aflibercept‚جƒIپ[ƒvƒ“ƒ‰ƒxƒ‹–³چىˆ×‰»‘وII‘ٹژژŒ± (AFFIRMژژŒ±) ‚ھچs‚ي‚ꂽ18)پBژه—v•]‰؟چ€–ع‚ج12ƒ•ŒژPFS‚حmFOLFOX6ŒQ (117—ل) 21.2%پAmFOLFOX6 + AfliberceptŒQ (119—ل) 25.8%‚إ‚ ‚èپAPFS’†‰›’l‚ح‚»‚ꂼ‚ê8.77ƒ•ŒژپA8.48ƒ•ŒژپA‘tŒّ—¦‚ح45.9%پA49.1%‚إ‚ ‚ء‚½پBŒQٹش”نٹr‚·‚éژژŒ±ƒfƒUƒCƒ“‚إ‚ح‚ب‚¢‚à‚ج‚جپAAflibercept•¹—p‚ة‚و‚é–¾‚ç‚©‚بڈمڈو‚¹Œّ‰ت‚ح”F‚ك‚ç‚ê‚ب‚©‚ء‚½پB
پ@2012”N8ŒژپA•ؤچ‘FDA‚حL-OHP‚ة‚و‚é‘Oژ،—أ—ً‚ج‚ ‚é‘ه’°ٹà‚ً‘خڈغ‚ةAflibercept‚ًFOLFIRI‚ئ‚ج•¹—p‚إڈ³”F‚µ‚½پB–{–M‚إ‚حپA‘ه’°ٹàٹùژ،—أ—ل‚ً‘خڈغ‚ةFOLFIRI + Aflibercept—أ–@‚ج‘وI‘ٹژژŒ±‚ھچs‚ي‚êپAگ„ڈ§—p—ت‚حٹCٹO‚ئ“¯‚¶4mg/kg‚ئŒˆ’肳‚ê‚ؤ‚¢‚é19)پBŒ»چفپA’†چ‘پE‘نکpپEƒVƒ“ƒKƒ|پ[ƒ‹‚إچs‚ي‚ê‚ؤ‚¢‚éL-OHPƒxپ[ƒX‚ج‰»ٹw—أ–@‚ة•s‰‚ئ‚ب‚ء‚½ٹ³ژز‚ً‘خڈغ‚ئ‚µ‚½VELOURژژŒ±‚ئ“¯—l‚جƒfƒUƒCƒ“‚ج‘وIII‘ٹژژŒ± (AFLAMEژژŒ±) ‚ةژQ‰ء‚µ‚ؤ‚¢‚é20)پB
پ@Ramucirumab‚حVEGFR-2‚جچ×–EٹOƒhƒپƒCƒ“‚ة“ءˆظ“I‚ةŒ‹چ‡‚·‚éٹ®‘SƒqƒgŒ^IgG1ƒ‚ƒmƒNƒچپ[ƒiƒ‹چR‘ج‚إ‚ ‚éپBVEGFR-2‚جƒٹƒKƒ“ƒh‚ة‚حپAVEGF-A‚¾‚¯‚إ‚ب‚VEGF-C‚âVEGF-D‚à’m‚ç‚ê‚ؤ‚¨‚èپA‚»‚ê‚ç‚جƒٹƒKƒ“ƒh‚ئVEGFR-2‚جŒ‹چ‡‚ً‘jٹQ‚·‚邱‚ئ‚إپAٹà‚جŒŒٹاگVگ¶‚ً—}گ§‚µچRژîل‡Œّ‰ت‚ً”ٹِ‚·‚éپB“–ڈ‰پAƒ}ƒEƒXƒqƒgƒLƒپƒ‰Œ^چR‘ج (IMC-1121) ‚ئ‚µ‚ؤٹJ”‚³‚ꂽ‚ھپA”¼گ”‚جٹ³ژز‚إƒqƒgچRƒLƒپƒ‰چR‘ج‚ھڈoŒ»‚µ‚½‚½‚كٹJ”’†ژ~‚ئ‚ب‚èپA‚»‚جŒمٹ®‘Sƒqƒg‰»چR‘ج (IMC-1121B) ‚ة‰ü—ا‚³‚ê‚ؤ—صڈ°ٹJ”‚ھچs‚ي‚ê‚ؤ‚¢‚é128)پB“ïژ،گ«ŒإŒ`ٹà‚ً‘خڈغ‚ئ‚µ‚½Ramucirumab’Pچـ—أ–@‚ج‘وI‘ٹ—p—ت‘Q‘ژژŒ±‚إ‚حپA37گl‚جٹ³ژز‚ة‘خ‚µ‚ؤ—p—ت2پ`16mg/kgپAڈT1‰ٌ‚إ“ٹ—^‚³‚ꂽ129)پBŒ‹‰تپA16mg/kgŒQ‚إ2—ل‚ةDLT (گ[•”گأ–¬ŒŒگًڈاپAچ‚ŒŒˆ³) ‚ھ”F‚ك‚ç‚êپAMTD‚ح13mg/kg‚ةŒˆ’肳‚ꂽپB‘ھ’è‰آ”\•a•د‚ً—L‚·‚é27—ل’†پAPR‚ً4—ل (ˆفٹàپAژq‹{•½ٹٹ‹ط“÷ژîپA—‘‘ƒٹàپAƒپƒ‰ƒmپ[ƒ}) ‚ة”F‚كپA‘ه’°ٹà2—ل‚ًٹـ‚ق7—ل‚إ6ƒ•Œژˆبڈم‚جSD‚ھ“¾‚ç‚ꂽپB‚»‚جŒمپA6-10mg/kg‚جٹuڈT“ٹ—^–@‚ج‘وI‘ٹژژŒ±‚ھچs‚ي‚ê (ڈعچׂبŒ‹‰ت‚ح–¢Œِ•\) پAŒ»چف‚إ‚ح8mg/kgٹuڈT“ٹ—^‚إٹJ”‚ھگi‚ٌ‚إ‚¢‚éپB
پ@–{–M‚إ‚àپA5-FU + L-OHP + Bevacizumab—أ–@‚ة•s‰‚ئ‚ب‚ء‚½‘ه’°ٹà‚ً‘خڈغ‚ةپAFOLFIRI—أ–@ (CPT-11‚ح180mg/m2) ‚ئRamucirumab 8mg/kgٹuڈT“ٹ—^‚ج ‘وIb‘ٹژژŒ±‚ھچs‚ي‚ꂽ130)پBDLT‚ح6—ل’†1—ل (grade 2‚ج’`”’”A‚ئgrade 4‚جچD’†‹…Œ¸ڈ‚ة‚و‚é2ڈTٹشˆبڈم‚ج“ٹ—^‰„ٹْ) ‚ة”F‚ك‚é‚ج‚ف‚إ‚ ‚ء‚½پBŒ»چفپA5-FU + L-OHP + Bevacizumab—أ–@‚ة•s‰‚ئ‚ب‚ء‚½‘ه’°ٹà‚ً‘خڈغ‚ئ‚µ‚ؤFOLFIRI + Ramucirumab—أ–@‚ئFOLFIRI—أ–@‚ً”نٹr‚·‚é‘وIII‘ٹژژŒ± (14T-MC-JVBBژژŒ±) ‚ھپA–{–M‚àژQ‰ء‚·‚éƒOƒچپ[ƒoƒ‹ژژŒ±‚ئ‚µ‚ؤچs‚ي‚ê‚ؤ‚¢‚éپB
پ@Vatalanib‚حVEGFR-1پAVEGFR-2پAVEGFR-3‚ً•W“I‚ئ‚µ‚½ƒ`ƒچƒVƒ“ƒLƒiپ[ƒ[‘jٹQچـ‚إ‚ ‚èپAin vitro‚إ‚ج50%‘jٹQ”Z“x (IC50) ‚ح‚»‚ꂼ‚ê77nMپA37nMپA270nM‚ئ•ٌچگ‚³‚ê‚ؤ‚¢‚é21)پB‘ه’°ٹàڈ‰‰ٌژ،—أ—ل‚ئ‚µ‚ؤFOLFOX4‚ئ•¹—p‚·‚é‘وIb‘ٹ—p—ت‘Q‘ژژŒ± (35—ل) ‚إ‚حپAVatalanib‚ةٹضکA‚·‚éDLT‚ئ‚µ‚ؤپA‚ك‚ـ‚¢پA‰^“®ژ¸’²‚ً”F‚كپAگ„ڈ§“ٹ—^—ت‚ح1,250mg/day 1“ْ1‰ٌ“ٹ—^‚ئŒˆ’肳‚ꂽ22)پB‚ـ‚½پAژ،—أŒّ‰ت‚ةٹض‚·‚é’Tچُ“I‚بŒں“¢‚ة‚¨‚¢‚ؤ‘tŒّ—¦48.6%پAPFS’†‰›’l11.4ƒ•Œژ‚ئ—اچD‚بŒ‹‰ت‚ًژ¦‚µ‚½‚½‚كپA‘وII‘ٹژژŒ±‚جŒ‹‰ت‚ً‘ز‚½‚¸پA‘ه’°ٹàڈ‰‰ٌژ،—أ—ل‚ً‘خڈغ‚ةFOLFOX4 + placebo—أ–@‚ئFOLFOX4 + Vatalanib—أ–@‚ً”نٹr‚·‚é‘وIII‘ٹژژŒ± (CONFIRM-1ژژŒ±) ‚ھچs‚ي‚ꂽ23)پB2003”N2Œژ‚©‚ç2004”N5Œژ‚ـ‚إ‚جٹْٹش‚ة1,168—ل‚ھ“oک^‚³‚êپAژه—v•]‰؟چ€–ع‚جPFS’†‰›’l‚حplaceboŒQ7.6ƒ•ŒژپAVatalanibŒQ7.7ƒ•Œژ (HR=0.88, 95% CI: 0.74-1.03, p=0.118)پAOS’†‰›’l‚حplaceboŒQ20.5ƒ•ŒژپAVatalanibŒQ21.4ƒ•Œژ (HR=1.08, 95% CI: 0.94-1.24, p=0.260) ‚ئپA‚ئ‚à‚ةچ·‚ً”F‚ك‚ب‚©‚ء‚½پBGrade 3/4‚ج—LٹQژ–ڈغ‚ئ‚µ‚ؤپAچ‚ŒŒˆ³ (6.8% vs. 23.0%)پA‚ك‚ـ‚¢ (2.3% vs. 7.4%)پA”xچاگًڈا (1.7% vs. 5.7%) ‚ب‚ا‚ھVatalanibŒQ‚إ—Lˆس‚ة‘½‚”F‚ك‚ç‚ꂽپB‚ـ‚½پADI (dose intensity) ‚ج’†‰›’l‚حپAL-OHP (65.8% vs. 60.6%)پA5-FU bolus’چژث (69.4% vs. 62.9%)پA5-FUژ‘±’چژث (70.6% vs. 64.4%) ‚¢‚¸‚ê‚àVatalanibŒQ‚إ’ل‚¢ŒXŒü‚ة‚ ‚ء‚½پB‚ب‚¨پA’Tچُ“IŒں“¢‚إ‚حپAژ،—أ‘OLDHچ‚’lŒQ (ژ{گفٹîڈ€ڈمŒہ‚ج1.5”{ˆبڈم) ‚ة‚¨‚¢‚ؤVatalanibŒQ‚إPFS‚ھ‰„’·‚·‚éŒXŒü‚ة‚ ‚ء‚½ (5.8ƒ•Œژ vs. 7.7ƒ•Œژ, HR=0.67, 95% CI: 0.49-0.91, p=0.009)پB
پ@‚³‚ç‚ةپA‘Oژ،—أ—ً‚ج‚ ‚é‘ه’°ٹàٹ³ژز855—ل‚ً‘خڈغ‚ةپAFOLFOX4 + placebo—أ–@‚ئFOLFOX4 + Vatalanib—أ–@‚ئ‚ً”نٹr‚µ‚½‘وIII‘ٹژژŒ± (CONFIRM-2ژژŒ±) ‚إ‚àپAژه—v•]‰؟چ€–ع‚جOS’†‰›’l‚حplaceboŒQ11.9ƒ•ŒژپAVatalanibŒQ13.1ƒ•Œژ‚ئڈمڈو‚¹Œّ‰ت‚ً”F‚ك‚ب‚©‚ء‚½ (HR=1.00, 95% CI: 0.87-1.16, p=0.957) 24)پBPFS’†‰›’l‚حplaceboŒQ4.2ƒ•ŒژپAVatalanibŒQ5.6ƒ•Œژ‚ئVatalanibŒQ‚إ—اچD‚بŒ‹‰ت‚ًژ¦‚µ (HR=0.83, 95% CI: 0.71-0.96, p=0.013)پAژ،—أ‘OLDHچ‚’lŒQ (ژ{گفٹîڈ€ڈمŒہ‚ج1.5”{ˆبڈم) ‚إ‚حپA‚و‚èچ·‚ھ‘ه‚«‚©‚ء‚½ (HR=0.63, 95% CI: 0.48-0.83, p<0.001)پB
پ@ˆبڈم‚©‚çپA‘ه’°ٹà‚ة‘خ‚·‚éFOLFOX4 + Vatalanib•¹—p—أ–@‚حپAژه—v•]‰؟چ€–ع‚ً’Bگ¬‚إ‚«‚¸پAٹJ”’†ژ~‚ئ‚ب‚ء‚½پB‚»‚ج—vˆِ‚ئ‚µ‚ؤپAVatalanib‚ج”¼Œ¸ٹْ‚ح4.6ژٹش‚ئ•ٌچگ‚³‚ê‚ؤ‚¢‚é‚ة‚à‚©‚©‚ي‚炸1“ْ1‰ٌ“ٹ—^‚ئ‚µ‚½‚±‚ئپA“إگ«‚ھڈمڈو‚¹‚³‚ê‚邽‚كFOLFOX4‚ج—p—ت‚ھ’ل‰؛‚·‚邱‚ئپAFOLFOX4‚ض‚جVEGFR‘jٹQ—أ–@‚حڈمڈو‚¹Œّ‰ت‚ھڈ¬‚³‚¢‚©‚à‚µ‚ê‚ب‚¢‚±‚ئپA‚ب‚ا‚ھگ„ژ@‚³‚ê‚ؤ‚¢‚éپBچRVEGFچR‘ج–ٍ‚إ‚ ‚éBevacizumab‚جگ¬Œ÷‚ھ‚ ‚èپAVEGFR‚ة‘خ‚·‚é’ل•ھژqگ«•ھژq•W“IچRٹàچـ‚ض‚جٹْ‘ز‚ھ‚ ‚ء‚½‚¾‚¯‚ةژc”O‚بŒ‹‰ت‚إ‚ ‚ء‚½پB
پ@Cediranib‚حژه‚ةVEGFR-1پAVEGFR-2پAVEGFR-3‚ً•W“I‚ئ‚µ‚½ƒ`ƒچƒVƒ“ƒLƒiپ[ƒ[‘jٹQچـ‚إ‚ ‚èپAin vitro‚إ‚جIC50‚ح‚»‚ꂼ‚ê5nMپA1nM–¢–پA3nM‚ئ’ل”Z“x‚إ‘jٹQŒّ‰ت‚ھ”F‚ك‚ç‚ê‚ؤ‚¢‚é25)پB‚ـ‚½پAc-KITپAPDGFR-ƒ؟پAPDGFR-ƒہپAFGFR1‚ة‘خ‚µ‚ؤ‚à’ل”Z“x‚إ‘jٹQŒّ‰ت‚ًژ‚آ‚ئ‚³‚ê‚éپB“ïژ،گ«ŒإŒ`ٹàٹ³ژز36—ل‚ً‘خڈغ‚ةچs‚ي‚ꂽCediranib’Pچـ‚ج—p—ت‘Q‘‘وI‘ٹژژŒ±‚ھچs‚ي‚ꂽ‚ھپAژه‚بDLT‚حچ‚ŒŒˆ³‚إ‚ ‚èپAMTD‚ح45mg/day 1“ْ1‰ٌ“ٹ—^‚ئŒˆ’肳‚ꂽ26)پB‘ه’°ٹà‚جڈ‰‰ٌژ،—أ—ل‚ً‘خڈغ‚ئ‚µ‚ؤچs‚ي‚ꂽmFOLFOX6 + Cediranib•¹—p—أ–@‚ج‘وI‘ٹژژŒ±‚إ‚حپA’تڈي—ت‚جmFOLFOX6‚ئCediranib (30mgپA45mg) ‚ج•¹—p‚ھژژ‚ف‚ç‚êپA30mg‚إ‚ج1—ل‚ة‚ج‚فDLT (grade 3‚ج‰؛—ں) ‚ھ”F‚ك‚ç‚ꂽ27)پBˆê•ûپA•Wڈ€ژ،—أ‚ة•s‰‚ئ‚ب‚ء‚½ٹ³ژز‚ً‘خڈغ‚ئ‚µ‚½mFOLFOX6 + Cediranib‚ج‘وI‘ٹژژŒ±28)‚إ‚حپA30mgŒQ‚ة‚¨‚¢‚ؤ5—ل’†2—ل‚إDLT‚ج”Œ» (grade 3‚ج‰؛—ںپA”وکJ) ‚ً”F‚كپAMTD‚ح20mg‚ئŒˆ’肳‚ꂽپB
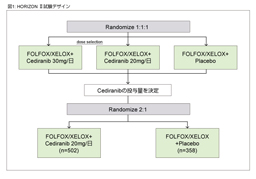
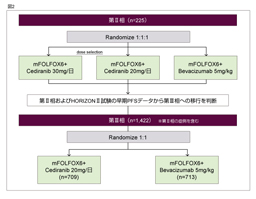
پ@‚±‚ê‚ç‚جŒ‹‰ت‚ً‚à‚ئ‚ةپAڈ‰‰ٌژ،—أ—ل‚ً‘خڈغ‚ةFOLFOX/CapOX + placebo—أ–@‚ئFOLFOX/CapOX + Cediranib—أ–@‚ئ‚ً”نٹr‚·‚é‘وIII‘ٹژژŒ± (HORIZON IIژژŒ±)پA‚ب‚ç‚ر‚ةFOLFOX6 + Bevacizumab—أ–@‚ئFOLFOX6 + Cediranib—أ–@‚ئ‚ً”نٹr‚·‚é‘وII/III‘ٹژژŒ± (HORIZON IIIژژŒ±) ‚ھچs‚ي‚ꂽپBژژŒ±ٹJژnژ‚ة‚حپA—¼ژژŒ±‚ةCediranib 20mgŒQ‚ئ30mgŒQ‚جژژŒ±ژ،—أŒQ‚ھگف’肳‚ê‚ؤ‚¢‚½‚ھپA—¼ژژŒ±‚ب‚ç‚ر‚ةٹùژ،—أ—ل‚ة‘خ‚·‚é–³چىˆ×‰»‘وII‘ٹژژŒ± (HORIZON IژژŒ±) ‚ًچ‡‚ي‚¹‚½’†ٹش‰ًگحŒم‚حپA20mgŒQ‚ج‚ف‚إژژŒ±‚ھŒp‘±‚³‚ꂽپBHORIZON IIژژŒ±29)‚إ‚حPFS‚ئOS‚ھ‚ئ‚à‚ةژه—v•]‰؟چ€–ع‚ئ‚µ‚ؤگف’肳‚ê‚ؤ‚¢‚½‚ھپAPFS’†‰›’l‚حplaceboŒQ8.3ƒ•ŒژپACediranib 20mgŒQ8.6ƒ•Œژ‚ئپACediranib 20mgŒQ‚إ—Lˆسچ·‚ً”F‚ك‚½‚à‚ج‚ج (HR=0.84, 95% CI: 0.73-0.98, p=0.0121)پAOS’†‰›’l‚حplaceboŒQ18.9ƒ•ŒژپACediranib 20mgŒQ19.7ƒ•Œژ‚إ‚ ‚èپA—Lˆسچ·‚ً”F‚ك‚ب‚©‚ء‚½ (HR=0.94, 95% CI: 0.79-1.12, p=0.5707)پBˆê•ûپAHORIZON IIIژژŒ± 30)‚حپAژه—v•]‰؟چ€–ع‚إ‚ ‚éPFS’†‰›’l‚ھBevacizumabŒQ10.3ƒ•ŒژپACediranibŒQ9.9ƒ•Œژ‚إ‚ ‚èپABevacizumabŒQ‚ة‘خ‚·‚é”ٌ—ٍگ«‚ًژ¦‚¹‚ب‚©‚ء‚½ (HR=1.10, 95% CI: 0.97-1.25)پB—LٹQژ–ڈغ‚حgrade 3/4‚ج‰؛—ںپA”وکJپAچD’†‹…Œ¸ڈپAچ‚ŒŒˆ³پAŒŒڈ¬”آŒ¸ڈ‚ج•p“x‚ھCediranibŒQ‚إ—Lˆس‚ة‘½‚پAژ،—أٹJژn‚©‚ç6ƒ•Œژژ“_‚ة‚¨‚¯‚éL-OHPپA5-FU‚جDI‚حCediranibŒQ‚إ’ل‚©‚ء‚½پB
پ@ˆبڈم‚ج—¼ژژŒ±‚جŒ‹‰ت‚©‚çپACediranib‚ح‘ه’°ٹà‚إ‚ج—صڈ°ٹJ”‚ھ’†ژ~‚³‚ꂽپBFOLFOX + Cediranib—أ–@‚حFOLFOX + placebo—أ–@‚ئ”نٹr‚µ‚ؤPFS‚ج‰„’·‚حژ¦‚µ‚½‚±‚ئ‚©‚çپAˆê’è‚جچRژîل‡Œّ‰ت‚ح—L‚·‚é‚ئچl‚¦‚ç‚ê‚é‚ھپA‰؛—ں‚ب‚ا‚ج—LٹQژ–ڈغ‚ج‘‹‚ھ–â‘è‚ئ‚ب‚ء‚½پB‚ب‚¨پA–{–M‚إچs‚ي‚ꂽmFOLFOX6‚ةCediranib (20mgپA30mg)پAplacebo‚ً•¹—p‚·‚é3ŒQ”نٹr‚ج‘وII‘ٹژژŒ±31)‚ة‚¨‚¢‚ؤ‚àپAplaceboŒQ‚ئ”نٹr‚µ‚ؤ20mgŒQ‚حPFS‚ھ—اچD‚إ‚ ‚ء‚½‚ھ30mgŒQ‚إ‚ح‹t‚ة‚â‚â—ٍ‚éŒXŒü‚إ‚ ‚ء‚½پB
پ@Brivanib‚حVEGFR-2پAVEGFR-3‚ً•W“I‚ئ‚µ‚½ƒ`ƒچƒVƒ“ƒLƒiپ[ƒ[‘jٹQچـ‚إ‚ ‚èپAin vitro‚إ‚جIC50‚ح‚»‚ꂼ‚ê23nM, 10nM‚ئ’ل”Z“x‚إ‘jٹQŒّ‰ت‚ھ”F‚ك‚ç‚ê‚é32)پB‚ـ‚½پAFGFR-1پAFGFR-2پAFGFR-3‚ة‘خ‚µ‚ؤ‚à‘jٹQŒّ‰ت‚ًژ‚؟پAin vitro‚إ‚جIC 50‚ح150nMپA125nMپA68nM‚ئ•ٌچگ‚³‚ê‚ؤ‚¢‚éپBŒإŒ`ٹà‚ً‘خڈغ‚ئ‚µ‚½Brivanib‚ج‘وI‘ٹژژŒ±‚إ‚حپA180پ`1,000mg‚ة‚ؤŒں“¢‚ھچs‚ي‚ꂽ‚ھپA1,000mg/day“à•ŒQ‚ج4—ل’†2—ل‚ةDLT‚ً”F‚كپAMTD‚ح800mg/day‚ئŒˆ’肳‚ꂽ33)پB800mg‚إچs‚ي‚ꂽٹg‘هƒRƒzپ[ƒg‚إ‚حپA•p“x‚جچ‚‚¢grade 3/4‚ج—LٹQژ–ڈغ‚حپA”وکJپAچ‚ŒŒˆ³پA‰؛—ںپA‚ك‚ـ‚¢پAٹج‹@”\ڈلٹQ‚إ‚ ‚ء‚½پBٹî‘bژہŒ±‚ة‚¨‚¢‚ؤEGFRŒoکH‚ئVEGFRŒoکH‚ج‘jٹQ–ٍ‚ً•¹—p‚·‚é‚ئ‘ٹڈو“I‚بچRژîل‡Œّ‰ت‚ھ”F‚ك‚ç‚ê‚é‚ئ‚¢‚¤•ٌچگ‚ھ‚ ‚ء‚½‚½‚كپACetuximab‚ئBrivanib‚ج•¹—p‚ة‚و‚é—p—ت‘Q‘‘وI‘ٹژژŒ±‚ھچs‚ي‚ꂽŒ‹‰تپA’تڈي—ت‚جCetuximab‚ئBrivanib’Pچـ—أ–@‚ة‚¨‚¯‚éMTD‚ج800mg‚ة‚و‚镹—p‚إ‚àMTD‚ة’B‚µ‚ب‚©‚ء‚½34)پBCetuximab + Brivanib (800mg) —أ–@‚ھچs‚ي‚ꂽ51—ل‚ة‚¨‚¯‚é—LٹQژ–ڈغ‚ج•p“x‚ح”畆ڈلٹQ27.5%پA”وکJ58.8%پA‰؛—ں35.3%پAگH—~•sگU37.3%پAASTڈمڈ¸37.3%‚ب‚ا‚إ‚ ‚ء‚½‚ھپAgrade 3/4‚ج—LٹQژ–ڈغ‚حڈ‚ب‚پA—LŒّگ«‚ةٹض‚µ‚ؤ‚حپAKRAS–ىگ¶Œ^25—ل‚إ‚جPFS’†‰›’l‚ھ7.2ƒ•Œژ‚ئٹْ‘ز‚إ‚«‚éگ¬گر‚إ‚ ‚ء‚½پB
پ@‚±‚جŒ‹‰ت‚ًژَ‚¯پAL-OHPپACPT-11•s‰‚جKRAS–ىگ¶Œ^‘ه’°ٹà‚ً‘خڈغ‚ةCetuximab + placebo—أ–@‚ئCetuximab + Brivanib—أ–@‚ئ‚ً”نٹr‚·‚é‘وIII‘ٹژژŒ± (NCIC/AGITG CO.20ژژŒ±) ‚ھچs‚ي‚ꂽ35)پBŒ‹‰تپAPFS’†‰›’l‚حplaceboŒQ3.4ƒ•Œژ‚ة‘خ‚µ‚ؤBrivanibŒQ4.8ƒ•Œژ‚ئBrivanibŒQ‚إ—Lˆس‚ة—اچD‚إ‚ ‚ء‚½‚ھ (HR=0.74, 95% CI: 0.64- 0.86, p<0.0001)پAژه—v•]‰؟چ€–ع‚إ‚ ‚éOS‚حپA’†‰›’l‚ھplaceboŒQ8.2ƒ•ŒژپABrivanibŒQ8.9ƒ•Œژ‚ئ—¼ŒQ‚ة—Lˆسچ·‚ً”F‚ك‚ب‚©‚ء‚½ (HR=0.89, 95% CI: 0.77-1.03, p=0.13)پBGrade 3ˆبڈم‚ج”ٌŒŒ‰t“إگ«‚حپA”وکJ (11% vs. 27%)پAچ‚ŒŒˆ³ (1% vs. 11%)پA‰؛—ں (3% vs. 8%)پAALTڈمڈ¸ (5% vs. 22%)پA”گ] (5% vs. 10%) ‚ھBrivanibŒQ‚إ‘½‚پADI‚ً90%ˆبڈمˆغژ‚إ‚«‚½ٹ„چ‡‚حپACetuximab‚ھ72% vs. 43%پAplacebo/Brivanib‚ھ86% vs. 47%‚ئپABrivanibŒQ‚إڈ‚ب‚©‚ء‚½پB
پ@Brivanib‚حCetuximab—أ–@‚ئ•¹—p‚·‚邱‚ئ‚ة‚و‚è‘tŒّ—¦پAPFS‚ج‰ü‘P‚ًژ¦‚µ‚½‚±‚ئ‚©‚çˆê’è‚جچRژîل‡Œّ‰ت‚ًژ‚آ‚ئچl‚¦‚ç‚ê‚é‚ھپAOS‚ج‰„’·‚ًژ¦‚·‚±‚ئ‚ح‚إ‚«‚ب‚©‚ء‚½پB‘وI‘ٹژژŒ±‚ئ‘وIII‘ٹژژŒ±‚ة‚¨‚¯‚égrade 3ˆبڈم‚ج”ٌŒŒ‰t“إگ«‚حپA”وکJ (12.9% vs. 27%)پA”çگ] (3.2% vs. 10%)پA‰؛—ں (1.6% vs. 8%)پAچ‚ŒŒˆ³ (1.6% vs. 11%) ‚إ‚ ‚èپA‘وI‘ٹژژŒ± (62—ل) ‚ة‚ح320mg (6—ل) پA600mg (5—ل) ‚ھٹـ‚ـ‚ê‚é‚à‚ج‚جپA‘وIII‘ٹژژŒ±‚إ‚ج—LٹQژ–ڈغ•p“x‚ھچ‚‚¢ŒXŒü‚ھ‚ف‚ç‚ꂽپB‚µ‚½‚ھ‚ء‚ؤپA‘وI‘ٹژژŒ±‚إ‚ح“إگ«‚ھ‰كڈ¬•]‰؟‚³‚ê‚ؤ‚¢‚½پA‚ـ‚½‚ح‘وIII‘ٹژژŒ±‚إ PS 2‚جڈا—ل‚ھ–ٌ10%ٹـ‚ـ‚ê‚ؤ‚¢‚½“™‚جˆل‚¢‚ھپA‘وIII‘ٹژژŒ±‚جŒ‹‰ت‚ة‰e‹؟‚ً‹y‚ع‚µ‚½‰آ”\گ«‚ھ‚ ‚éپB
پ@Sunitinib‚حVEGFR-2پAPDGFRپAc-KITپAFLT3پAFGFR1‚ب‚ا‚جƒ`ƒچƒVƒ“ƒLƒiپ[ƒ[‚جATPŒ‹چ‡•”ˆت‚ة‹£چ‡“I‚ة‘jٹQچى—p‚ً—L‚·‚éپA•ھژq—ت398.48‚ج’ل•ھژqگ«•ھژq•W“IچRٹàچـ‚إ‚ ‚é36)پBŒإŒ`ٹà‚ً‘خڈغ‚ةچs‚ي‚ꂽ‚¢‚‚آ‚©‚ج‘وI‘ٹژژŒ±‚جŒ‹‰تپAکA‘±“ٹ—^–@‚إ‚ح’~گدگ«‚ھ”F‚ك‚ç‚êپA4ڈT“à•2ڈT‹x–ٍ‚ج“ٹ—^–@‚ة‚ؤGISTپAگtچ×–Eٹà‚إچRژîل‡Œّ‰ت‚ھ”F‚ك‚ç‚ꂽ‚±‚ئ‚©‚çپA“¯—l‚جƒXƒPƒWƒ…پ[ƒ‹‚إ‚»‚جŒم‚ج—صڈ°ژژŒ±‚ھچs‚ي‚ê‚ؤ‚¢‚é37)پB‚ـ‚½پA75mg/day‚ة‚ؤDLT (grade 3‚ج”وکJپAچ‚ŒŒˆ³) ‚ھ”F‚ك‚ç‚êپA‘O—صڈ°ژژŒ±‚إ50mg/day‚ة‚ؤPDGFRپAVEGFR-2‘jٹQ‚ة•K—v‚ئ‚³‚ꂽŒŒ’†”Z“x‚ة’B‚µ‚½‚±‚ئ‚©‚çپA50mg/day‚ھگ„ڈ§—p—ت‚ئŒˆ’肳‚ꂽپB‚»‚µ‚ؤپA2003”N‚©‚ç5-FUپACPT-11پAL-OHP‚ًٹـ‚قژ،—أ—ً‚ج‚ ‚é‘ه’°ٹàٹ³ژز84—ل‚ً‘خڈغ‚ةSunitinib’Pچـ—أ–@‚ج‘وII‘ٹژژŒ±‚ھچs‚ي‚ꂽŒ‹‰تپA‘tŒّ—¦‚ح1.2%پA22ڈTˆبڈم‚جSDژ‘±‚ح15.9%‚إ‚ ‚ء‚½38)پBBevacizumab–¢ژg—p—ل (40—ل) ‚ئBevacizumabژg—p—ل (42—ل) ‚ئ‚ج”نٹr‚إ‚حپA22ڈTˆتڈم‚جSDژ‘±‚ح–¢ژg—p—ل27.5%پAژg—p—ل4.8%‚ئپA–¢ژg—p—ل‚إ’·ٹْ‚ج•aگ¨ˆہ’è‚ھ“¾‚ç‚ê‚éŒXŒü‚ھŒ©‚ç‚ꂽ‚ھپA’Pچـ‚إ‚جŒّ‰ت‚ح–R‚µ‚¢‚ئچl‚¦‚ç‚ꂽپBGrade 3/4‚ج—LٹQژ–ڈغ‚حپA”وکJ (14.6%)پA‰؛—ں (11.0%)پAŒŒڈ¬”آگ”Œ¸ڈ (8.5%)پAچ‚ŒŒˆ³ (6.1%) “™‚ھ”F‚ك‚ç‚ꂽ‚ھپA”E—e‰آ”\‚إ‚ ‚ء‚½پB
پ@‚»‚±‚إپA‘ه’°ٹàڈ‰‰ٌژ،—أ—ل‚ً‘خڈغ‚ةmFOLFOX6‚¨‚و‚رFOLFIRI‚ض‚جSunitinib‚ج•¹—p—أ–@‚ھژژ‚ف‚ç‚ꂽپBmFOLFOX6 + Bevacizumab—أ–@‚ئmFOLFOX6 + Sunitinib (37.5mg/dayپA4ڈT“à•2ڈT‹x–ٍ) —أ–@‚ئ‚ً”نٹr‚·‚é–³چىˆ×‰»‘وII‘ٹژژŒ±‚إ‚حپAPFS’†‰›’l‚حBevacizumabŒQ11.2ƒ•ŒژپASunitinibŒQ9.1ƒ•Œژ‚إ‚ ‚è (HR=1.598, p=0.96)پASunitinibŒQ‚ج—D‰zگ«‚حژ¦‚³‚ê‚ب‚©‚ء‚½39)پBGrade 3/4‚ج—LٹQژ–ڈغ‚حچD’†‹…Œ¸ڈ (22% vs. 66%)پAŒŒڈ¬”آگ”Œ¸ڈ (3% vs. 34%) ‚ھSunitinibŒQ‚إ•p“x‚ھچ‚‚©‚ء‚½پB‚ـ‚½پAFOLFIRI + placebo—أ–@‚ئFOLFIRI + Sunitinib (37.5mg/dayپA4ڈT“à•2ڈT‹x–ٍ) —أ–@‚ئ‚ً”نٹr‚µ‚½‘وIII‘ٹژژŒ±‚إ‚حپAژه—v•]‰؟چ€–ع‚جPFS‚إ—LŒّگ«‚ھژ¦‚³‚ꂸ’†ٹش‰ًگح‚إ’†ژ~‚ئ‚ب‚ء‚ؤ‚¢‚é40)پBPFS’†‰›’l‚حplaceboŒQ8.4ƒ•ŒژپASunitinibŒQ7.8ƒ•Œژ (HR=1.095, p=0.8072)پAOS’†‰›’l‚حplaceboŒQ19.8ƒ•ŒژپASunitinibŒQ20.3ƒ•Œژ‚إ‚ ‚ء‚½ (HR=1.171, p=0.9163)پBGrade 3/4‚ج—LٹQژ–ڈغ‚حپAچD’†‹…Œ¸ڈ (30% vs. 68%)پA‰؛—ں (8% vs. 16%)پAŒŒڈ¬”آگ”Œ¸ڈ (1% vs. 11%) ‚ب‚ا‚ة‚¨‚¢‚ؤSunitinibŒQ‚إ•p“x‚ھچ‚‚©‚ء‚½پB
پ@‘ه’°ٹà‚ة‘خ‚·‚éSunitinib‚ح’Pچـ‚إ‚حŒّ‰ت‚ھ–R‚µ‚پAچ×–EڈٹQگ«چRٹàچـ‚ئ‚ج•¹—p‚إ‚حŒŒ‹…Œ¸ڈ‚ب‚ا“إگ«‚ج‘‹‚ھ”F‚ك‚ç‚ꂽپB–{–M‚إچs‚ي‚ꂽ‘وI‘ٹژژŒ±‚إ‚àپAmFOLFOX6 + Sunitinib (37.5mg/dayپA4ڈT“à•2ڈT‹x–ٍ) —أ–@‚إ‚جٹe–ٍچـ‚جDI‚حپASunitinib‚ھ50.4%پAL-OHP‚ھ56.3%پA5-FU bolus/infusional‚ھ39.2%/53.1%‚ئ•ٌچگ‚³‚ê‚ؤ‚¨‚è41)پAڈ‰‰ٌژ،—أ‚ة‚¨‚¯‚ékey drug‚إ‚ ‚é5-FUپAL-OHP‚ج“ٹ—^—ت‚ھ‹ة’[‚ةڈ‚ب‚‚ب‚ء‚ؤ‚µ‚ـ‚¤‚±‚ئ‚ھ‘ه‚«‚ب–â‘è“_‚ئچl‚¦‚ç‚ê‚éپB
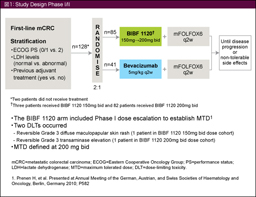
پ@BIBF1120‚حVEGFR1-3پAPDGFR-ƒ؟/ƒہپAFGFR1-3‚ً•W“I‚ئ‚µ‚½’ل•ھژqگ«•ھژq•W“IچRٹàچـ‚إ‚ ‚éپB‘ه’°ٹà‚جڈ‰‰ٌژ،—أ‚ئ‚µ‚ؤmFOLFOX6 + Bevacizumab—أ–@‚ئmFOLFOX6 + BIBF1120—أ–@‚ئ‚ً”نٹr‚·‚é–³چىˆ×‰»‘وII‘ٹژژŒ±‚إ‚حپA9ƒ•ŒژPFS‚ھBevacizumabŒQ69%پABIBF1120ŒQ63%‚ئ“¯“™‚إ‚ ‚ء‚½42)پB—LٹQژ–ڈغ‚حBIBF1120ŒQ‚إŒy“x‚إ‚ ‚èپAڈء‰»ٹاگْچE‚ًٹـ‚قڈء‰»ٹيŒn‚جڈd“ؤ‚ب—LٹQژ–ڈغ”گ¶ٹ„چ‡‚àBIBF1120ŒQ‚إڈ‚ب‚©‚ء‚½ (29.3% vs. 11.8%)پBŒ»چفپACPT-11ƒxپ[ƒXƒŒƒWƒپƒ“Œم‚ج2nd-line‚ة‚¨‚¢‚ؤmFOLFOX6 + placebo—أ–@‚ئmFOLFOX6 + BIBF1120—أ–@‚ئ‚ً”نٹr‚·‚é–³چىˆ×‰»‘وII‘ٹژژŒ± (TRICC-CژژŒ±) ‚ھچs‚ي‚ê‚ؤ‚¢‚é43)پB
پ@Axitinib (AG-013736) ‚حVEGFR1-3‚ج‘I‘ً“I‚بƒ`ƒچƒVƒ“ƒLƒiپ[ƒ[‘jٹQچـ‚إ‚ ‚èپAŒ»چفپAگtچ×–Eٹà‚ة‘خ‚µ‚ؤ•ؤچ‘FDAپA“ْ–{‚إ‚جڈ³”F‚ھ“¾‚ç‚ê‚ؤ‚¢‚éپB‘ه’°ٹà‚جڈ‰‰ٌژ،—أ—ل‚ً‘خڈغ‚ةپAmFOLFOX6‚ة‘خ‚·‚éBevacizumab•¹—pŒQپAAxitinib•¹—pŒQپABevacizumab + Axitinib•¹—pŒQ‚ً”نٹr‚µ‚½–³چىˆ×‰»”نٹr‘وII‘ٹژژŒ±‚إ‚حپA‘tŒّ—¦‚حBevacizumab•¹—pŒQ49%پAAxitinib•¹—pŒQ29%پABevacizumab + Axitinib•¹—pŒQ39%‚إ‚ ‚èپAPFS’†‰›’l‚ح‚»‚ꂼ‚ê350“ْپA315“ْپA377“ْ‚ئپAAxitinib‚ج—LŒّگ«‚حژ¦‚³‚ê‚ب‚©‚ء‚½44)پB
پ@Pazopanib‚حVEGFR1-3پAPDGFR-ƒ؟/ƒہپAc-KIT‚ة‘خ‚·‚é‘jٹQچـ‚إ‚ ‚èپAˆ«گ«“î•”ژîل‡‚ة‘خ‚µ‚ؤ2012”Nڈ³”F‚³‚ê‚ؤ‚¢‚éپB‘ه’°ٹà‚ة‘خ‚µ‚ؤ‚حپAڈ‰‰ٌژ،—أ‚ة‚¨‚¯‚éFOLFOX6/XELOX‚ئ‚ج•¹—p—أ–@‚â45)پA2nd-line‚ة‚¨‚¯‚éCPT-11 + Cetuximab‚ئ‚ج•¹—p—أ–@‚ة‚آ‚¢‚ؤ‚ج‘وI‘ٹژژŒ±‚ج•ٌچگ‚ھ‚ ‚é‚à‚ج‚ج46)پA‚»‚جŒم‚جٹJ”‚حچs‚ي‚ê‚ؤ‚¢‚ب‚¢‚و‚¤‚إ‚ ‚éپB
پ@Tivozanib (AV-951) ‚حVEGFR1-3‚ج‘I‘ً“I‘jٹQچـ‚إ‚ ‚éپBŒ»چفپAڈ‰‰ٌژ،—أ—ل‚ً‘خڈغ‚ةmFOLFOX6 + Bevacizumab—أ–@‚ئmFOLFOX6 + Tivozanib—أ–@‚ج–³چىˆ×‰»”نٹr‘وII‘ٹژژŒ±‚ھچs‚ي‚ê‚ؤ‚¢‚é47)پB
پ@Vandetanib (ZD6474) ‚حVEGFR-2 (IC50 40nM)پAVEGFR-3 (110nM)پARET (130nM)پAEGFR (500nM) ‚ب‚ا‚جƒ}ƒ‹ƒ`ƒLƒiپ[ƒ[‘jٹQچـ‚إ‚ ‚é48)پBCPT-11•s‰—ل‚ً‘خڈغ‚ئ‚µ‚½mFOLFOX6 پ} Vandetanib (100mgپA300mg) 49)پAL-OHP•s‰—ل‚ً‘خڈغ‚ئ‚µ‚½FOLFIRI پ} Vandetanib (100mgپA300mg) ‚ج–³چىˆ×‰»‘وII‘ٹژژŒ±‚ھچs‚ي‚ꂽ‚ھ50)پA‚¢‚¸‚ê‚à–¾‚ç‚©‚بڈمڈو‚¹Œّ‰ت‚ح”F‚ك‚ç‚ê‚ب‚©‚ء‚½پB
پ@Linifanib (ABT-869) ‚حپAVEGFR-2 (IC50 8nM)پAFLT-1 (3nM)پAPDGFR-ƒ؟ (29nM)پACSF-1R (5nM) ‚ب‚ا‚ً•W“I‚ئ‚µ‚½ƒ}ƒ‹ƒ`ƒLƒiپ[ƒ[‘jٹQچـ‚إ‚ ‚éپBmFOLFOX6 + Bevacizumab—أ–@‚ئپAmFOLFOX6 + Linifanib—أ–@‚ئ‚ً”نٹr‚µ‚½–³چىˆ×‰»‘وII‘ٹژژŒ±‚ھچs‚ي‚ꂽ‚ھپABevacizumabŒQ‚ًڈم‰ٌ‚éŒّ‰ت‚ح”F‚ك‚ç‚ê‚ب‚©‚ء‚½51)پB
GI cancer-net
ڈء‰»ٹيٹàژ،—أ‚جچLڈê