コンパニオン診断薬の開発に必要な諸要件
土原:これまで現状の問題点についてお話いただきましたが、これからは今後の臨床開発における課題について進めていきたいと思います。まず、どうすれば厳格な質保証のもとでコンパニオン診断薬を開発できるのか、登先生から解説していただきます。
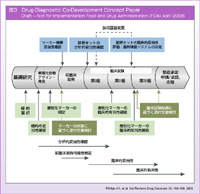
登:2005年に米国FDAが出したconcept paperでは、コンパニオン診断薬と治療薬は同時開発、同時承認という方向性が打ち出されています (図3)。ここでは、前臨床開発の時点で層別化マーカーを同定し、マーカー検査の妥当性を確認したうえで診断キットの分析的妥当性確認を行い、臨床試験へと移っていきます。そして、臨床試験を実施しながら診断キットのプラットフォームについても考慮し、第II相試験、あるいは第III相試験の段階では、既に最終的なシステムが決定され、第III相試験の終了時点で製造承認申請を行います。
米国ではこの仕組みにより、2011年8月にはALK 融合遺伝子陽性肺癌に対するCrizotinib、そしてBRAF V600E変異陽性メラノーマに対するVemurafenibが、いずれもコンパニオン診断薬と同時に承認されました。Vemurafenibのコンパニオン診断薬であるcobas® 4800 BRAF V600 Mutation Testの開発が開始されたのは、前臨床段階の2005年まで遡ります11)。2007年に治験機器に対する一部規則の適用免除 (IDE: investigational device exemption) の認可を受け、2009年より第II相試験、および第III相試験に使用され、市販前承認 (PMA: Premarket Approval) されました。
一方、慢性骨髄性白血病 (CML) では、T315I変異陽性例に対してOmacetaxineが開発されましたが、第II相試験、第III相試験においてT315I変異を検査した施設で、検査方法がそれぞれ異なりました。この状況では承認を得られないため、コンパニオン診断薬の開発は進めずに方針転換し、最終的には先行する2剤に抵抗性か、忍容性のない患者に限定することで承認されました。つまり、しっかりとコンパニオン診断薬が開発されなかったために、治療薬の適応が変わってしまったわけです。この例は、コンパニオン診断薬の開発が創薬において非常に重要であることを示しています。
日本でも治療薬とコンパニオン診断薬を同時開発、同時承認する仕組みをつくる必要があります。そのためにも、治療薬とコンパニオン診断薬の開発を行う企業が歩調を合わせて開発を進め、第II相試験の段階ではコンパニオン診断薬を使用できるような体制をつくらなければなりません。
土原:ありがとうございます。コンパニオン診断薬は新規の治療薬に対して1対1対応でしか承認が得られないのでしょうか。
登:コンパニオン診断薬とは、文字通り1対1で治療薬の開発と一緒に進むものという考えになっています。
土原:つまり、マルチプレックス診断は、コンパニオン診断薬が何種類も発売された後に、効率的に診断するための後発品のようなイメージになるのですね。

登:現在のところはそうなります。例えばマルチプレックスで検査することにより、「KRAS に変異があったため、この薬剤は使えません」で終わるのではなく、「別の変異があるので、別の薬剤が使えます」となれば、患者にも大きなベネフィットがあります。同じ遺伝子検査を受けるにしても、複数のオプションを示せるのであれば検査に前向きになれると思います。
吉野:西尾先生が進められているNGSによるマルチプレックス診断は、臨床の場ではどの段階で使用されるとお考えでしょうか。
西尾:国際臨床試験への参加も視野に入れているため、プレスクリーニングを想定しています。実際に稀少変異例を対象にした臨床試験では、NGSを使用している施設を適格とするプロトコールも増えています。したがって、製薬企業のバイオマーカーに対する考え方も変わり、マルチプレックスへと移っていくのではないかと思います。
土原:米国では治療薬とコンパニオン診断薬の同時開発で成功を収めましたが、さらに次のパラダイムにシフトしつつあるということですね。つまり、日本でコンパニオン診断薬開発の仕組みを作る前に、世界は一足飛びに次の段階へ移行してしまう恐れがあります。この現状を把握し、現状に合致した制度をつくらねばなりません。
先進医療とコンパニオン診断薬
土原:日本における新規技術の導入におけるユニークなシステムとして先進医療がありますが、この制度について教えていただけますでしょうか。
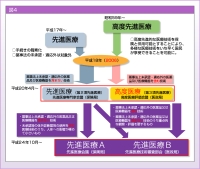
登:混合診療を避けるために昭和59年から始まった高度先進医療制度は、平成20年に再編成され、第2項先進医療 (先進医療) と第3項先進医療 (高度医療) とに分けられました (図4)。両者の違いは薬事承認がされているか否かで、未承認のものは第3項先進医療として扱われます。また、第2項先進医療は保険局、第3項先進医療は医政局の所掌で、第3項先進医療は先進医療専門家会議の前に高度医療評価会議により技術評価としての妥当性の確認がされるため、手続きに時間がかかります。吉野先生からご紹介いただいたKRAS 検査は、診断薬が未承認のため本来は第3項先進医療にあたるのですが、遺伝子検査は第2項先進医療として承認されるケースが多く、判定基準が曖昧でした。
平成24年10月からは先進医療Aと先進医療Bに分けられ、未承認の体外診断薬でも人体への影響が極めて小さいものに関しては、先進医療Aに区分されるようになりました。臨床検査側から見ると比較的規制が緩くなったと言え、home-brew assayなどを導入しやすくなったと思います。
ただ、保険局と医政局の縦割りは続いており、先進医療Aは保険局、先進医療Bは医政局の所掌のままです。また、先進医療などの評価療養は、もともと混合診療を避けるための制度なので、イノベーション (技術革新) などとは無縁です。我々が今議論していることはまさしくイノベーションなので、どのように保険診療のなかに組み込んでいくかを国として考えていく必要があります。
土原:科学的で精度の高い診断方法を日常診療に早期導入するためにも、先進医療の仕組み自体は有意義ですが、日本発の新技術を開発し、世界に広げていくためには、現状のままでは厳しく、制度そのものを見直していくよう働きかけることが大事ですね。
|
||
| ▲ このページのトップへ |